Ning-Xin Gu, Yang Liu. Genetic evaluations of island populations of Rhesus Macaque (Macaca mulatta) in China: Implications for conservation management[J]. Zoological Research: Diversity and Conservation, 2024, 1(1): 75-78. DOI: 10.24272/j.issn.2097-3772.2023.070
Citation:
Ning-Xin Gu, Yang Liu. Genetic evaluations of island populations of Rhesus Macaque (Macaca mulatta) in China: Implications for conservation management[J]. Zoological Research: Diversity and Conservation, 2024, 1(1): 75-78. DOI: 10.24272/j.issn.2097-3772.2023.070
Ning-Xin Gu, Yang Liu. Genetic evaluations of island populations of Rhesus Macaque (Macaca mulatta) in China: Implications for conservation management[J]. Zoological Research: Diversity and Conservation, 2024, 1(1): 75-78. DOI: 10.24272/j.issn.2097-3772.2023.070
Citation:
Ning-Xin Gu, Yang Liu. Genetic evaluations of island populations of Rhesus Macaque (Macaca mulatta) in China: Implications for conservation management[J]. Zoological Research: Diversity and Conservation, 2024, 1(1): 75-78. DOI: 10.24272/j.issn.2097-3772.2023.070
School of Life Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510275, China
2.
Shenzhen Research Institute, Sun Yat-sen University, Shenzhen, Guangdong 518057, China
3.
School of Ecology, Shenzhen Campus, Sun Yat-sen University, Shenzhen, Guangdong 518107, China
Funds: This research was supported by Shenzhen Municipal Science & Technology Innovation Committee (JCYJ20180504170040910) and Urban Administration & Law Enforcement Bureau of Shenzhen Municipality (201802)
The macaques belongs to the genus Macaca, consisting of at least 23 species (Roos et al., 2019). Among all congeners, rhesus macaque (M. mulatta) is regarded as the widest distributed non-human primate species in the world. Its native range spans in East Asia, northern part of Southeast Asia and Indian subcontinent (Liu et al., 2018). Listed as “Least Concern” on the IUCN Red List, this species is locally threatened due to habitat loss and degradation in China and Thailand (Lu et al., 2018). Nevertheless, pet release resulting in hybridization with other congeners (e.g., rhesus macaque × crab-eating macaque (M. fascicularis)) was documented in Hong Kong SAR, China (Wong & Ni, 2000), threatening genetic integrity of wild populations.
Chinese populations of rhesus macaque are mainly distributed in southern China. Previous population genomics revealed that it contains five genetic lineages in China (Liu et al., 2018). Island macaque populations live on Hainan Island, Wanshan islands of Pearl River Delta, as well as islands adjacent to Hong Kong (Figure 1A). These populations were unique insular inhabitants with higher population density and reproductive success, yet a lower flock size and a smaller home range as compared with continental counterparts (Zhang et al., 2016). The taxonomic status of Hainan Island populations belongs to the subspecies brevicaudus supported by the recent population genomic work (Liu et al., 2018). The populations of Wanshan Island and Kowloon Island of Hong Kong may belong to this subspecies. However, their status remains contentious. Apart from natural colonization from China during the Quaternary (Zong et al., 2009), alternative hypothesis includes anthropogenic-mediated introduction (Chu et al., 2019). However, the genetic profile of island macaque populations has not yet been examined.
Figure
1.
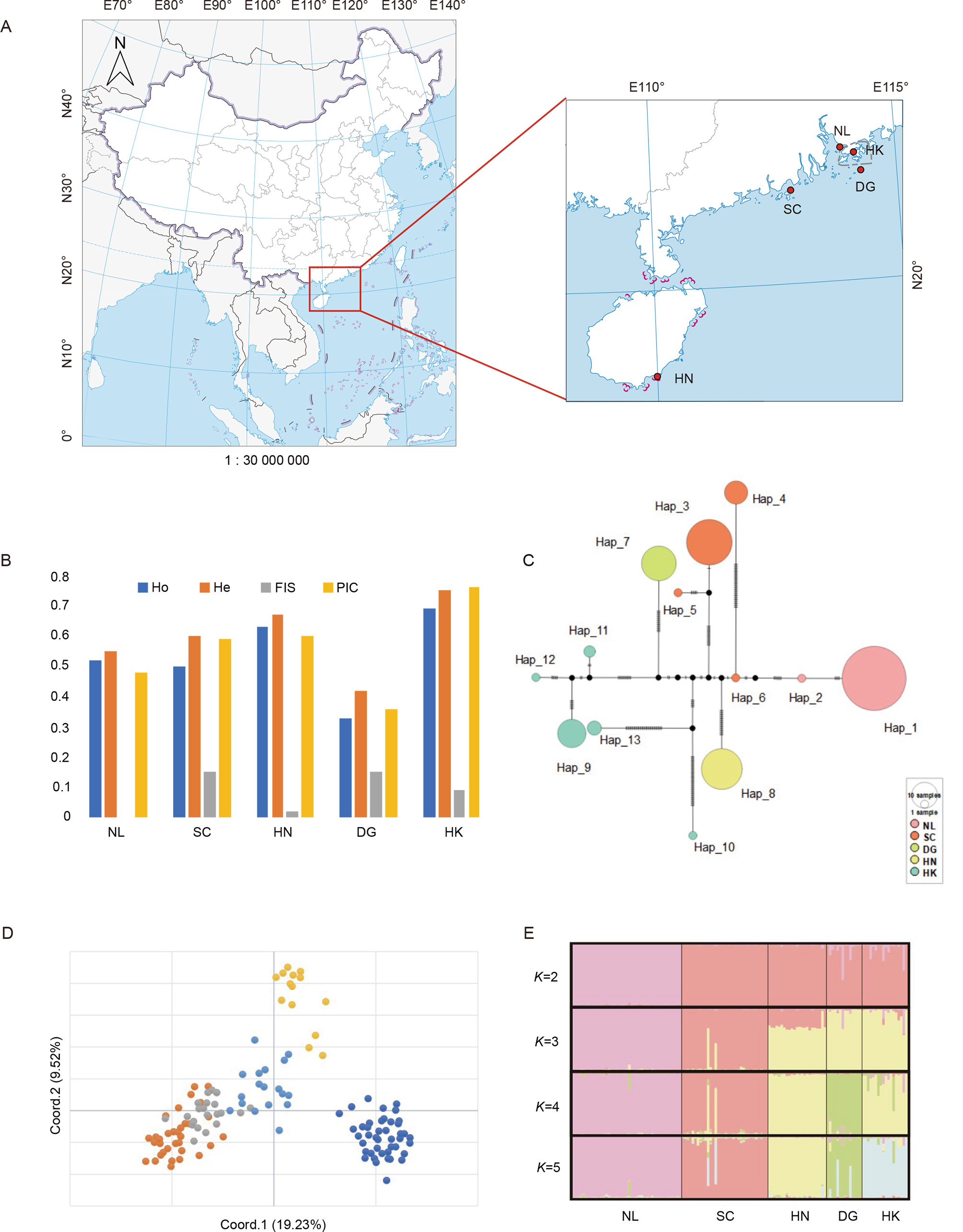
Sample localities, genetic diversity, and population structuring of island populations of rhesus macaque (Macaca mulatta) in southern China
A: Sampling localities of offshore populations of rhesus macaque in southern China. NL: Neilingding Island; SC: Shangchuan Island; HN: Nanwan Peninsula in Hainan Island; DG: Dangan Island; HK: Kowloon Island of Hong Kong. B: Genetic diversity indices in five study populations were estimated by ten microsatellites of observed heterozygosity (Ho), expected heterozygosity (He), inbreeding coefficient (FIS) and polymorphic information content (PIC). C: Unrooted median-joining networks depicted detailed genetic distances of haplotypes out of five study populations based upon 835 bp of mtDNA fragment. Size of circle was proportional to the number of individuals with a particular haplotype. D: Plots of the first two coordinates from principal coordinates analysis based upon Euclidian distances between multi-locus microsatellite genotypes. E: Bayesian clustering analysis with STRUCTURE based upon 10 microsatellite genotypes of 132 individuals from five populations. Plots of population assignment of each individuals in relative to each of given genetic cluster was represented by the length of each line under K=2‒5.
Here wild rhesus macaques were sampled from five offshore populations in southern China, namely Neilingding Island (NL), Shangchuan Island (SC), Dangan Island (DG), Kowloon Island of Hong Kong (HK) and Nanwan of Hainan Island (HN) respectively (Figure 1A). Size and conservation status of each population varied widely (Supplementary Table S1). For the first time, conservation genetic analysis was performed on rhesus macaques in islands of Guangdong and Hainan. Diverse variations were genotyped by both maternally inherited mitochondrial DNA (mtDNA) and polymorphic autosomal microsatellite markers. The authors attempted to assess their genetic diversity, genetic differentiation, and some demographic signatures such as genetic drift and gene flow among island populations. A total of 181 individual samples were collected, including 180 fresh fecal samples and one tissue sample from a corpse. We successively genotyped 167 samples of an 835 bp mtDNA fragment and 132 samples at 10 microsatellite loci (Supplementary Table S2). Unsuccessful samples were due to low DNA quality, failure in PCRs or too many missing values at microsatellite genotypes (>3 loci). The levels of genetic diversity varied among localities. For mtDNA dataset, both DG and HN had only one haplotype representing samples (18, 25 individuals) with decedents from a single matrilineal ancestor (Supplementary Table S3). Genetic diversity was slightly higher for NL since two haplotypes were identified. The populations of SC and HK had relatively higher haplotype and nucleotide diversities (Supplementary Table S3). For microsatellites, deviations from Hardy–Weinberg equilibrium (HWE) occurred at all loci when analyzing all populations together (Supplementary Table S2), though no violation of HWE was detected at each locus per population. It hinted at population sub-structuring within samples. There was no evidence of genotypic disequilibrium after Bonferroni correction. Among the above five populations, DG offered the lowest genetic diversity in all estimated indices while other populations had a moderate level of genetic diversity (Figure 1B). Three populations showed no sign or an extremely low level of inbreeding. Yet DG and SC had larger inbreeding coefficients (Supplementary Table S3).
For mtDNA data, combing of a dataset of our 167 samples, 296 published sequences and one outgroup (M. sylvanus), 277 unique haplotypes out of a total of 464 sequences were classified by 110 polymorphic sites. The phylogenetic relationships of 277 unique haplotypes were examined with Bayesian Inference (Supplementary Figure S1). Almost all individuals from the above five study populations belonged to a well-supported mitochondrial haplogroups from eastern China. However, one exception from Hong Kong was nested in the western Chinese haplogroup, consisting of published sequences from wild populations in western China (Wu et al., 2013).
The haplotype networks provided finer descriptions, showing no shared haplotypes among populations. Haplotypes were distantly correlated with mutational steps of 10 to >20. Genetic diversity was extremely low at mtDNA in two populations of DG and HN since a single haplotype has been detected in each population (Supplementary Table S3). In contrast, HK/SC had a comparably higher level of haplotypic diversity. In HK population, though haplotypes (Hap_9\11\12) were closely correlated with each other with a few steps. Also a few haplotypes were distantly correlated with each other. For instance, one HK haplotype (Hap_10 in Supplementary Figure S1) belonged to the western Chinese haplogroup (Figure 1C). Two SC haplotypes were also distantly related. Such a pattern caused high haplotypic diversity in these two populations.
For microsatellites, three methods were applied for describing population subdivision among five populations. Pairwise genetic differentiations indicated that Fst values among populations comparisons were significantly above zero, hinting at marked genetic barrier among populations (Supplementary Table S4). The pattern of population differentiation was further visualized by the first two coordinates from principal coordinate analysis (Figure 1D). Furthermore, Bayesian clustering analysis with STRUCTURE revealed two genetic clusters in all 10 replicates (peaking at Delta K=2), with SC, DG, HK and HN populations grouped together, NL formed one cluster (Figure 1E). However, there was also a small peak (Delta K=4) denoting potential substructures within these two major genetic clusters (Supplementary Figure S2). Then genetic components were plotted for examining four genetic clusters. NL, SC, and DG formed a genetic cluster while HK and HN joined another common cluster. There was congruence with the lowest Fst value between HK and HN (Supplementary Table S4). Interestingly, two individuals of SC seemed to share some genetic components from HN/HK. A few individuals in HK also revealed genetic footprint of introgression from DG/NL.
There was no strong evidence of a recent bottleneck at five study sites (Supplementary Table S5). All the results of Wilcoxon’s test under two-phase model (TPM) were not significant, except for DG population. Moreover, NL had a weak evidence of recent bottleneck, as suggested by the result of one tail for heterozygosity deficiency from Wilcoxon’s test under Stepwise Mutation Model (SMM). Furthermore, gene flow among five populations of rhesus macaque remained quite weak as revealed by Bayesass (Supplementary Table S6).
Mitochondrial genetic variations among five populations may be explained by three key factors of inheritance mode, sex-biased dispersal, and demographics. Firstly, the genotyped fragment spanned the control region of mtDNA. As a non-coding region, this mitogenomic locus had a faster substitutional rate probably due to balancing selection. This may partially explain distant mutational steps among haplotypes between populations (Figure 1C). Secondly, under a maternally inherited mode of mtDNA, the observed haplotypic pattern reflected no shared history from matrilineal line, meaning that these sites were colonized by different female ancestor(s) and evolved independently each. Indeed, such a premise might be also supported by sex-biased dispersal in rhesus macaques. Females tended not to leave their social groups and led a high extent of female philopatry in populations (Roos & Zinner, 2015). Hence, populations with low genetic diversity hinted at a limited number of female founders in those island populations of DG, NW, and HN. In contrast, populations of HK and SC with more alien founders were supported by greater genetic divergence among haplotypes.
Autosomal microsatellite loci are biparentally inherited polymorphic markers for implementing information offered by mtDNA. A moderate level of genetic diversity was detected in five populations genotyped by microsatellites. The most valuable information lied in that inbreeding co-efficient was significantly higher than random mating in SC/DG. This result was reasonable as sizes of these two populations were rather smaller (ca. 200–300 individuals) as compared with the remaining three (Supplementary Table S1). In particularly, the signal of historical population bottleneck was detected in DG population. Moreover, DG population offered genetic distinctiveness as suggested by PCA/STRUCTURE analysis (Figure 1D, E). Coupling with an extremely low mitochondrial diversity, DG population had a high priority for protection among these five populations.
Though geographically close to HK, NL population manifested some genetic distinctiveness from the other four populations through microsatellites (Figure 1D, E). Two non-mutually exclusive hypotheses helped to explain the observation pattern. Firstly, this distinct ancestral population might represent an independent colonization event at Neilingding Island. This scenario was possible since no mitochondrial haplotype was shared between NL and other populations. For genotyping individuals from any continental population, it was rather not likely to assign NL to any of geographically possible ancestral populations in China. Secondly, this population developed historical population bottleneck (Supplementary Table S5). Genetic drift fluctuated allele frequency and consequently manifested genetic divergence from other populations. These interacting processes could have shaped the evolutionary trajectory of NL population. It hinted that NL population should be managed independently. Fortunately, Neilingding Island has been a part of Neilingding-Futian National Nature Reserve and local macaque population is strictly protected. With a growing trend, this population shows no sign of degradation (Chu et al., 2019).
Though populations of HK and SC show deep divergence in mtDNA, together with HN, these three populations have relatively a lower level of divergence as shown in microsatellites. Thus ancestral populations of these three sites may be derived from more closely related ancestors, perhaps representing common ancestors from southern China. SC population manifested a sign of high inbreeding and marked genetic differentiation (when K=4 in STRUCTURE analysis, Figure 1E). A small population size and genetic drift may explain the observed pattern. Introducing male individuals may be a feasible solution for lowering the inbreeding level of this population.
It is worth noting that HK had the highest level of genetic diversity in both mtDNA and microsatellites. It may be explained by population admixture from different origins (Wong & Ni, 2000). For HN, though it contained only one mitochondrial haplotype, genetic diversity from paternal side may be high as shown in a high level of genetic diversity by microsatellites. Unlike other populations, only 25 individuals were sampled from HN population and it failed to fully represent the entire Hainan macaque population (Supplementary Table S1) due to a larger geographical coverage and a greater sample size (Zhang et al., 2016).
In conclusion, our results suggest substantial population genetic differentiation among five study populations. Founder effects, bottleneck, inbreeding and human-mediated colonization are probably the major factors for shaping their contrasting pattern of genetic diversity. Proper conservation practices, such as population monitoring, habitat restoration, supplementary feeding, predator removal, introducing male macaques should be employed to aid population augmentation and lower risk of inbreeding depression of island rhesus macaques in southern China. Nonetheless, no genotyped data from continental Guangdong region has hindered resolving subspecies affinity of these island populations. Combining complete sampling and population genomics hold a great promise in future studies to elucidate the origins and anthropogenic impacts on the population trajectories of unique island macaques.
ACKNOWLEDGEMENTS
The authors thank Peng Zhang, Zhi-Jin Liu, Guo-Ling Chen, Fan Da, Peng-Fei Fan, Hai-Sheng Jiang and Shi-Xiao Yu for their assistances regarding the funding, data collection and manuscript composing. We sincerely appreciate three anonymous reviewers for their insight-full comments on our paper.
AUTHORS’ CONTRIBUTIONS
N.X.G. and Y.L. designed the project. N.X.G. conducted sampling, molecular works and conducted analyses. N.X.G. and Y.L. composed the paper. All authors read and approved the final version of the manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ChuYMR, Zhan QJ, Yang Q, et al. 2019. Population dynamic and viability analysis of Rhesus Macaque (Macaca mulatta) in Neilingding Nature Reserve, Guangdong province. Chinese Journal of Wildlife, 40(2): 259−266. (in Chinese)
Liu ZJ, Tan XX, Orozco-ter Wengel P, et al. 2018. Population genomics of wild Chinese rhesus macaques reveals a dynamic demographic history and local adaptation, with implications for biomedical research. GigaScience, 7(9): giy106.
Lu JQ, Tian JD, Zhang P. 2018. Advances in ecological research regarding rhesus macaques (Macaca mulatta) in China. Acta Theriologica Sinica, 38(1): 74−84. (in Chinese)
Roos C, Kothe M, Alba DM, et al. 2019. The radiation of macaques out of Africa: evidence from mitogenome divergence times and the fossil record. Journal of Human Evolution, 133: 114−132. doi: 10.1016/j.jhevol.2019.05.017
Roos C, Zinner D. 2015. Diversity and evolutionary history of macaques with special focus on Macaca mulatta and Macaca fascicularis. In: Bluemel J, Korte S, Schenck E, et al. The Nonhuman Primate in Nonclinical Drug Development and Safety Assessment. Amsterdam: Elsevier, 3–16.
Wu SJ, Luo J, Li QQ, et al. 2013. Ecological genetics of Chinese rhesus macaque in response to mountain building: all things are not equal. PLoS One, 8(2): e55315. doi: 10.1371/journal.pone.0055315
Zhang P, Lyu MY, Wu CF, et al. 2016. Variation in body mass and morphological characters in Macaca mulatta brevicaudus from Hainan, China. American Journal of Primatology, 78(6): 679−698. doi: 10.1002/ajp.22534
Zong Y, Yim WWS, Yu F, et al. 2009. Late Quaternary environmental changes in the Pearl River mouth region, China. Quaternary International, 206(1-2): 35−45. doi: 10.1016/j.quaint.2008.10.012


 DownLoad:
DownLoad:


